Metagenomics Sequence Analysis
If you're interested in joint analysis of 16S and shotgun metagenomic datasets from the HMP, pairing up data from the same microbiome samples can initially seem tricky. The following tables join 16S dataset "SN" and "PSN" identifiers with metagenomic dataset "SRS" identifiers. The diagram below indicates how these sample IDs are related experimentally.
The DACC provides mapping files organized by 16S variable region (V), containing metadata and linking together all levels of ids:
Sample flow of the clinical phases is described in the following schematic:
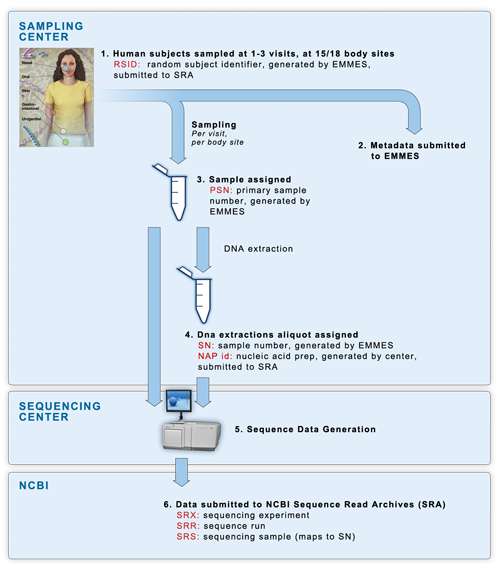
- (1) Samples were collected at 2 dedicated sampling centers (see Sample collection for more detail), with each human participant being sampled at 15 or 18 body sites over one to three visits.
- (2) All subject & sample metadata was submitted to EMMES, our partner in metadata storage and management. EMMES assigned each human subject a de-identified RSID (random subject identifier).
- (3) Every individual sample was assigned a PSN id, an EMMES-generated Primary Sample Number.
- (4) DNA was extracted from the primary samples using methods described in the Manual of Procedures, and assigned a NAP id (Nucleic Acid Preparation) generated by the sequencing centers. Extractions were divided into aliquots as necessary. Each aliquot was assigned an SN id, an EMMES-generated Sample Number. For the clinical pilot phase, every primary sample was sent to two sequencing centers to assess reproducibility of technical replicates across multiple institutions. Therefore there is a one-to-many relationship between PSN and SN ids. In the clinical pilot phases, a single PSN sample generated a single SN sample, which was sent to a single sequencing center, allowing for a one-to-one relationship between PSN and SN.
- (5) Sequencing centers performed sequencing on each SN sample, sequencing 16S variable regions 3-5 (V35) from every samples within the study, and variable regions 1-3 (V13) and variable regions 6-9 (V69) from subsets of samples. If the SN samples did not provide enough material for sequencing, the centers were sent the original PSN samples for re-extraction and sequencing.
- (6) All sequence data was submitted by the sequencing centers directly to NBCI's Sequence Read Archives (SRA) data repository. Centers submitted data using the RSID subject ids, and the NAP sample ids. Data was then organized at NCBI into their standardized levels of experiment accession (SRX ids), run id (SRR ids) and sample id (SRS ids).
Member Organizations



